1) molecular orbital theory

分子轨道理论
1.
The molecular orbital theory was utilized to investigate the crystal cluster model of NO adsorbed on TiO 2 and the change of band gap in the adsorption process.
采用程序升温热脱附 (TPD)实验方法测定了NO在TiO2 表面吸附后的脱附谱 ,利用分子轨道理论研究了TiO2吸附NO的原子簇模型及吸附前后的原子簇能级变化 。
2.
The clusters of the NO adsorption on the TiO2(110) were calculated with the molecular orbital theory of MOPAC and Gaussian, and the charge distribution and energy level of the TiO2(110) clusters were investigated.
利用MOPAC和GAUSSIAN分子轨道理论计算了在TiO2(110)表面上吸附NO分子的原子簇模型,电荷分布以及原子簇的能级,推断了NO在TiO2(110)表面吸附的稳定性。
3.
The relation between resonance theory and molecular orbital theory was illustrated by concrete examples.
通过实例说明了共振论与分子轨道理论的关系。
2) molecular orbit theory
分子轨道理论
1.
Using the self experience molecular orbit theory(complete neglect differential overlap)(CNDO/2), this paper calculates the bond length of Silicon Hydrogen in the Silane molecular system, works out a theory formula about the relationship between the extending vibration frequency and bond length of Silicon Hydrogen in Silane molecular system.
论文利用半经验分子轨道理论方法—全略微分重叠(CNDO/2)计算了硅烷类分子体系中的硅氢键长,拟合出硅烷类分子体系中硅氢伸张振动频率同硅氢键长关系的理论公式,利用该拟合公式计算了硅氢伸张振动频率,并同频率实验值进行了比较。
3) semi-empirical molecular orbital theory
半经验分子轨道理论
4) Polyhedral molecular orbital method
多面体分子轨道理论
5) Molecular orbital valence bond principle
分子轨道价键理论
6) molecular orbital-valence bond (MOVB)
分子轨道-价键理论
补充资料:分子轨道理论
| 分子轨道理论 molecular orbital theory 一种化学键理论。原子轨道理论对分子的自然推广。其基本观点是:物理上存在单个电子的自身,只受分子中的原子核和其他电子平均场的作用,以及泡利不相容原理的制约;数学上则企图将难解的多电子运动方程简化为单电子方程处理。因此,分子轨道理论是一种以单电子近似为基础的化学键理论。描写单电子行为的波函数称轨道(或轨函),所对应的单电子能量称能级。对任何分子,如果求得了它的系列分子轨道和能级,就可以像讨论原子结构那样讨论分子结构,并联系到分子性质的系统解释。有时,即便根据粗糙的计算方案所得到的部分近似分子轨道和能级,也能分析出很有用处的定性结果。 正如在原子轨道理论中,氢原子的严格解提供了进一步发展的理论模式,氢分子离子 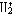 中,单个电子在固定核间距R 的双质子场中的波动 方程解,即氢分子离子的分子轨道是分子轨道理论进程中的基石。 中,单个电子在固定核间距R 的双质子场中的波动 方程解,即氢分子离子的分子轨道是分子轨道理论进程中的基石。  的分子轨道用符号σ、π、δ、…表征,对应于精 确解中的 量子数m=0,±1,±2 ,…,它描述相对于核间距R的轨道对称行为。此外,还需用g和u表征相对于分子中心反演的对称行为。综合起来, 的分子轨道用符号σ、π、δ、…表征,对应于精 确解中的 量子数m=0,±1,±2 ,…,它描述相对于核间距R的轨道对称行为。此外,还需用g和u表征相对于分子中心反演的对称行为。综合起来,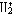 的分子轨道用σg 、σu、πg 、πu 、…等符号表征 ,借助精确求解固定核间距R 的波动方程获 得。其中两个能量最低的轨道为1σg和1σu,在能量E随R的变化曲线中,1σg能级有一极小值( R=2a0,a0 为玻尔半径),代表基态。1σu的行为不同 ,能量随R减 少而单调 上升,显示排斥态的本质。1σg和1σu也被称作成键轨道和反键轨道。作为一种近似处理 ,分子轨道可以当作原子轨道的线性组合 ,简写为LCAO,它的一般形式是: 的分子轨道用σg 、σu、πg 、πu 、…等符号表征 ,借助精确求解固定核间距R 的波动方程获 得。其中两个能量最低的轨道为1σg和1σu,在能量E随R的变化曲线中,1σg能级有一极小值( R=2a0,a0 为玻尔半径),代表基态。1σu的行为不同 ,能量随R减 少而单调 上升,显示排斥态的本质。1σg和1σu也被称作成键轨道和反键轨道。作为一种近似处理 ,分子轨道可以当作原子轨道的线性组合 ,简写为LCAO,它的一般形式是: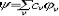 。式中ψ是分子轨道或轨函; 。式中ψ是分子轨道或轨函;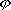 υ是属于各组成原子的原子轨道;cυ 是待定系数 ,由变分法确定。 υ是属于各组成原子的原子轨道;cυ 是待定系数 ,由变分法确定。用 LCAO近似来讨论任意双原子分子中,分属两个原子a和b的一对原子轨道 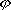 a和 a和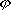 b( 能量分别为 Ea和Eb )形成分子轨道的最优条件,得到有效的成键作用决定于 3 个条件:①能量近似条件,即│Ea-Eb│愈小愈好。②最大重叠条件,即 b( 能量分别为 Ea和Eb )形成分子轨道的最优条件,得到有效的成键作用决定于 3 个条件:①能量近似条件,即│Ea-Eb│愈小愈好。②最大重叠条件,即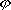 a和 a和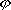 b的重叠愈大,成键作用可能愈大。③对称性条件。 b的重叠愈大,成键作用可能愈大。③对称性条件。可将上述概念和方法推广用于复杂的多原子分子,但不能精确求解,利用对称性分析(群论),可建立能级相关图、分子轨道对称守恒原理和休克分子轨道法等。进一步考虑电子间的排斥作用,建立了哈特里 - 福克方程自洽场分子轨道法以及多种组态相互作用分子轨道从头计算方法。 |
说明:补充资料仅用于学习参考,请勿用于其它任何用途。
参考词条