1) canonical variational transition state theory

正则变分过渡态理论
1.
The rate constants for the title reaction are calculated by canonical variational transition state theory(CVT) with small-curvature tunneling(SCT) correction in the temperature range 200 .
利用正则变分过渡态理论,结合小曲率隧道效应校正(CVT/SCT)方法计算了该反应的两个可行的反应通道在200 K~2 000 K温度范围内的速率常数。
2) improved canonical variational transition state (theory)
改进的正则变分过渡态理论
3) variational transition state theory
变分过渡态理论
1.
In this review we provide new application of variational transition state theory-inreaction at gas-solid interface ,with emphasis on dissociation absorption of gas-phase moleculeon the surface of face-centered cubic crystal.
提供了变分过渡态理论的新应用-用于气固界面上的反应,重点是气相分子在面心立方晶体上发生的解离吸附反应,给出了反应的物理图象,用新的势能面计算了H_2在Ni(100)面上的解离吸附的速率常数,结果与Truhlar的结果相符。
2.
A transplanted and reformed variational transition state theory is used to calculate the reaction rate constants for titled state-selected reactions, which pass some distinctly vibrational excited transition states.
本文用移植改造的变分过渡态理论,计算了标题中反应经过不同振动激发过渡态时的速度常数,发现给定相同的能量,过渡态的弯曲振动模式的激发比伸缩振动模式激发更有利于反应的进行,本文还分析了反应的动力学瓶颈区性质。
4) Transition state theory
过渡态理论
1.
In this paper,the rate constant and equilibrium constant of the crack reaction of methyl silicane were obtained with the transition state theory and quantum chemistry calculation.
本文采用过渡态理论和量子化学计算方法 ,首次计算得到了甲硅烷裂解反应的速率常数和平衡常数 ,并对结果进行了讨论。
2.
On the basis of the quantum chemistry theory and the transition state theory, the degradation reactions of 1,1,1,2-tetrafluoroethane (HFC-134a) and 1,1-difluoroethane (HFC-152a) in the atmosphere are investigated theoretically to reveal their mechanism and kinetics properties.
采用过渡态理论加Eckart隧道效应校正(TST/Eckart)在200-1000K温度范围内计算了反应速率与温度变化的关系式分别为HFC-134a + OH:k(T) =1。
6) Variational transition state
变分过渡态
1.
In this paper,the reaction of NH 3 molecule and ground state NH radical is studied by the “direct dynamics” method of variational transition state theory,which uses the information on electronic structure energy and energy derivatives calculated ab initio along the minium energy path.
利用变分过渡态理论的“直接动力学”方法对NH3分子和基态NH(X3∑)自由基的反应进行了理论研究;利用从头算计算得到反应体系的电子结构能和能量梯度等信息,计算了200~2500K温度范围该反应的速率常数和穿透系数,分析了影响隧道效应和反应速率常数的一些因素。
2.
Furthermore, the dynamical properties along the reaction path and CVT (canonical variational theory) rate constants with correction of tunneling effect are investigated by reaction path Hamiltonian theory and variational transition state theory.
用从头算UHF/6-31G方法计算CH3+OH→CH2+H2O的反应途径,在此基础上计算沿反应途径的动态学性质及正则变分过渡态理论的速率常数,同时进行隧道效应的校正。
补充资料:过渡态理论
| 过渡态理论 transition-state theory 研究有机反应中由反应物到产物的过程中过渡态的理论。过渡态理论是1935年由A.G.埃文斯和M.波拉尼提出的。 过渡态和活性中间体 有机反应可分为一步反应(协同反应)和分步反应两大类。其中,一步反应只有过渡态,没有活性中间体;而分步反应既有过渡态,又有活性中间体 。活性中间体并非就是过渡态,两者不可混淆。例如氯代叔丁烷的水解反应分两步进行: (CH3)3CCl→(CH3)3C++Cl- (CH3)3C++H2O→(CH3)3COH+H+其间出现两个过渡态、一个中间体——正碳离子(CH3)3C+。这两步反应分别经过两个势垒ΔE1和ΔE2,过渡态1——(CH3)3C…Cl和过渡态2——(CH3)3C…OH2分别出现于每步的势能顶峰处,而活性中间体处于两峰之间的凹谷处。活性中间体与两个过渡态的结构和性质相近,但不相同。一般,中间体很活泼,寿命很短,但比过渡态要稳定,故可用各种现代物理化学方法测定其结构。两个过渡态之间的中间体的势能越低,则中间体越稳定。利用中间体结构和性能的知识 ,可大致推断出过渡态的结构、性能,以阐明反应机理。 反应速率决定步骤和产物决定步骤 从氯代叔丁烷水解反应来看,第一步反应的活化能比第二步反应大得多(ΔE1>ΔE2),因此第一步反应比第二步反应慢得多。第一步反应是总反应速率的关键,称为反应速率决定步骤,第一步的过渡态称为速率决定过渡态。 如果在上述的氯代叔丁烷水溶液中加入第二种比水的亲核性更强的试剂。例如叠氮负离子N3-,则反应的主要产物为叔丁基叠氮化物,而原有产物叔丁醇很少。这是由于水和N3-分别与叔丁基正碳离子反应并相互竞争。对于两者来说,第一步均形成叔丁基正碳离子,是速率决定步骤,第二步反应是竞争反应: 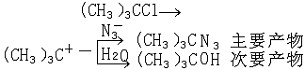 由于N3-的亲核性比水强,其第二步反应活化能更小,即ΔE2(H2O)>ΔE2(N3- ),(CH3)3C+与N3-反应要比水快 ,N3-在第二步反应的竞争中占了优势 ,故主要产物为(CH3)3CN3。由于第二步反应的势垒(ΔE2)比第一步(ΔE1)小得多,反应速率也高得多,是决定所得产物的步骤,因此称第二步反应为产物决定步骤。由此可见,如果知道一个有机反应中的中间体和过渡态的类型,并通过动力学方法确定其中的速率决定步骤和产物决定步骤,就能推知整个反应机理 。 活化熵的意义 由过渡态理论的基本公式ΔG≠ =ΔH≠-TΔS≠可以知道,反应活化熵ΔS≠ 对反应速率有一定的影响,通常其作用比活化焓ΔH≠要小,然而从反应活化熵的变化可以获得有关过渡态立体化学特征的知识。通常,若ΔS≠ 是负值,即熵值减少,则过渡态结构的有序性有所增加;反之,若ΔS≠ 是正值 ,则其有序性减少,ΔS≠ 负值过大不利于反应。此外,还必须综合比较ΔH≠ 和ΔS≠ 变化的相对大小对于活化自由能ΔG≠的影响。例如,两端为-OH和- COOH 基团的10个碳原子的直链化合物在关环成内酯时,其过渡态结构的有序性大为增加,熵值减少很多,即上述公式的第二项向正值方向变化,使ΔG≠ 向正值方向变化也大 ,不利于反应 ;而含3、4个碳原子的化合物在进行相应的关环反应时,虽然熵值负值很小,但ΔH≠正值很大,ΔS≠ 的变化难以抵消ΔH的不利作用,结果ΔG≠ 很高,反应也难以发生 。五、六元环的ΔH≠和ΔS≠综合作用最为有利,所以容易成环。 |
说明:补充资料仅用于学习参考,请勿用于其它任何用途。
参考词条