1) density functional theory

密度泛函
1.
Comparative studies on the structures of porphyrin(H_2P),using hartree-fock and density functional theory methods;
卟吩结构的HARTREE-FORCK和密度泛函研究比较
2.
Study of density functional theory on N-alkylpyridium cations and aluminium chloride anions;
烷基吡啶及氯化铝正负离子结构的密度泛函研究
3.
The density functional theory study on the fluorescent emission spectra of naphthaline derivatives;
萘类衍生物荧光发射光谱的密度泛函理论研究
2) DFT
密度泛函
1.
DFT Study of Asymmetrical Annular-Addition of Methylenecyclopropanes with 1,3-Dicarbonyl Compound Free Radicals;
亚甲基环丙烷与1,3-双羰基化合物自由基不对称环加成反应的密度泛函研究
2.
DFT study on heterobinuclear Cu(Ⅱ)-Co(Ⅱ) complex of N,N′-bis(3-carboxyl salicyl aminal ethylene) oxalamide;
N,N′-双(3-羧基水杨醛叉缩胺乙基)草酰胺Cu(Ⅱ)-Co(Ⅱ)异双核配合物的密度泛函研究
3.
The Synthesis and DFT Research of Tetraethyl 1,2-Diphenylethane-1,1,2,2-Tetracarboxylate;
1,2-二苯基-1,1,2,2-四乙氧基羰基乙烷的合成及密度泛函计算研究
3) density function
密度泛函
1.
Comparative investigation of the molecular electrostatic potential of fullerenes with density function theory and HF ab initio methods;
密度泛函理论和从头算方法对富勒烯分子静电势的比较研究
2.
Application of density functional theory for the study on IR harmonic vibration spectra of ammonia clusters (NH_3)_n(n=2~8);
氨团簇(NH_3)_n(n=2~8)红外振动光谱的密度泛函理论研究
3.
The mechanism of the addition reaction of disilyne and hydrogen has been studied by using ab initio calculation of quantum chemistry and density functional theory.
利用量子化学从头算和密度泛函理论(DFT),研究了硅炔和氢气分子加成生成甲硅烷基硅烯的反应机理。
4) density functional
密度泛函
1.
Through density functional calculations, the mechanism of Michael type addition was investigated with alkynyl carbene complex as the substrate and 3.
利用密度泛函理论研究了炔基钨卡宾为底物的Michael加成反应机理,发现吡唑上取代基团的增大可以导致第三步反应的活化能大于第一步,从而使反应的决速步骤由原来的第一步转变为第三步。
2.
Optimized and calculated geometric structure and infrared spectrum of benzoic acid molecule with density functional B3LYP method on 6-311G ** base cluster level,and compared the calculated infrared frequency of each conformation of benzoic acid with actually-measured infrared spectrum,thus determined molecula.
用密度泛函B3LYP方法在6-311G**基组水平上对苯甲酸分子的几何结构和红外光谱进行了优化和计算,并将计算所得的苯甲酸各构象的红外频率与其实测的红外光谱进行比较,确定了在晶体中苯甲酸的分子结构,同时在计算中给出了各种频率所对应的红外强度,对光谱进行了归属。
3.
Fourteen models of germanium clusters Ge11 have been designed by using molecular graphics software, and the geometry optimization and calculation on vibrational frequency been carried out by means of B3LYP density functional method.
用分子图形软件设计出多种Ge_11原子团簇模型,使用B3LYP密度泛函方法进行几何构型优化和振动频率计算,比较了14种同分异构体的总能量,得到了新的基态构型。
5) Density function theory
密度泛函
1.
Calculation of structure of alkylamine chloroaluminates molten salts by density function theory;
氯化烷基季铵盐离子液体结构性质的密度泛函计算
2.
A study on La(Ⅲ) doped anatase titanium dioxide by density function theory
La(Ⅲ)掺杂TiO_2密度泛函理论的研究
3.
Molecular structure and IR spectrum data of alkyllimidazolium chloride were calculated successfully by density function theory.
采用量子化学密度泛函的方法模拟计算了氯化烷基咪唑离子液体的分子结构和红外光谱,并用实测值考证了计算结果。
6) Density functional theory(DFT)
密度泛函(DFT)
补充资料:泛函
| 泛函 functional 定义于一般集合,取数值(实数值或复数值)的映射。又称泛函数。是微积分中函数概念的发展和拓广。例如,取非空集X={f:f为定义在[a,b]上的连续函数},映射F:X→R为F(f)= 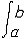 f(x)dx。F就是定义在集合X上的一个泛函。因此,泛函可通俗地看作以函数为自变元的函数,在很多情况下,泛函的定义域均为函数集。泛函概念的产生直接与变分法中的求极值问题有关。上面的例子还具有这样的性质: f(x)dx。F就是定义在集合X上的一个泛函。因此,泛函可通俗地看作以函数为自变元的函数,在很多情况下,泛函的定义域均为函数集。泛函概念的产生直接与变分法中的求极值问题有关。上面的例子还具有这样的性质: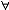 f,g∈X 和 f,g∈X 和 α,β∈R,有以下等式成立:F(αf+βg)= α,β∈R,有以下等式成立:F(αf+βg)= [αf(x)+βg(x)]dx= α [αf(x)+βg(x)]dx= α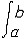 f(x)dx+β f(x)dx+β g(x)dx=αF(f)+βF(g),称这样的泛函为线性泛函。线性泛函是算子理论研究的主要对象之一,也是研究空间性质及结构的重要工具。因为利用线性连续泛函可导出对偶空间(由定义在X上的一切线性连续泛函组成的集合,并赋以自然的加法、数乘运算及范数)的概念,而空间X与其对偶空间 X*间的关系对于认识空间本身的性质和研究算子的性质都起着基础的作用。 g(x)dx=αF(f)+βF(g),称这样的泛函为线性泛函。线性泛函是算子理论研究的主要对象之一,也是研究空间性质及结构的重要工具。因为利用线性连续泛函可导出对偶空间(由定义在X上的一切线性连续泛函组成的集合,并赋以自然的加法、数乘运算及范数)的概念,而空间X与其对偶空间 X*间的关系对于认识空间本身的性质和研究算子的性质都起着基础的作用。除此之外,对应于多线性算子有多重线性泛函概念,例如在希尔伯特空间中,双线性泛函作为表示工具在处理问题时十分方便有效。 |
说明:补充资料仅用于学习参考,请勿用于其它任何用途。
参考词条